Whole exome sequencing (WES) targets the protein-coding regions of the genome and as it requires sequencing of only 1-2% of the genome, is much more cost-effective than sequencing the whole genome.
Source Genomics offers a flexible range of packages to suit your research needs. Utlilising the Nextera Flex for enrichment library protocol we can offer a range of different capture panels that best meet your project requirements.
Primarily we use the Agilent SureSelect Human All Exon V7 capture panel with other panels include Twist all Exon and IDT Exome Research panel, whilst we also provide the flexibility to incorporate other panels of choice including those specifically designed for your needs.
A free consultation is provided at the initiation stage of the project and an a dedicated Account Manager will be assigned to provide end-to-end support throughout the project and post data delivery.
Overview
Sequencing package details
Our standard package sequences to a depth of 100x raw coverage, with results using the SureSelect Human all exon panel providing a mean 27x coverage of >95% of targeted bases from ~5Gb of data - typically outperforming Agilent specifications.
Our standard package offering includes:
- Exome enrichment and library preparation
- Illumina 150bp PE sequencing – 100x raw coverage – 5Gb.
- Return of raw FastQ files via secure FTP or HDD
Optional extra services available:
- Additional sequencing depth
- DNA Extraction
- Bioinformatics analysis
The process
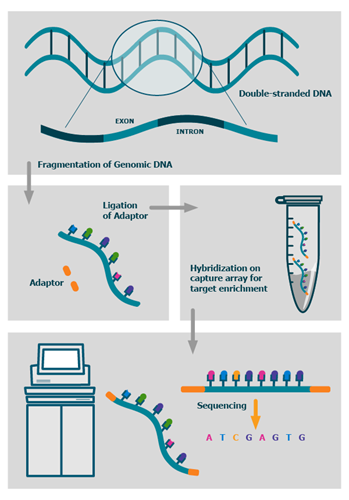
We also offer advanced BioIT variant analysis packages as well as fully bespoke BioIT solutions.
BioInformatics Variant Calling
Germline:
Investigation of germline single nucleotide polymorphisms (SNPs), insertions and deletions (INDELs) present in either whole genome or exome data.
This analysis uses Next Generation Sequencing data to identify any germline mutations by comparison to a reference sequence. Raw reads are trimmed, aligned and deduplicated, followed by variant calling, recalibrating, filtering and finally annotating variants with entries from relevant public databases such as dbSNP and ClinVar. You will receive raw (fastq) and aligned (bam) files for each sample, as well as VCF files listing all variants. A bioinformatics report will provide further detail about the tools used and alignment metrics and a variant quality stats.
Additionally, access to an interactive online viewing tool (Relyter) is available now.
Somatic:
Identification and quantification of somatic single nucleotide variants (SNVs), insertions and deletions (INDELs).
This analysis uses Next Generation Sequencing data from cancer samples to identify somatic mutations by comparison to either a reference sequence and/or a matched healthy sample. Raw reads are trimmed, aligned, deduplicated and somatic variants are called via local de-novo assembly of haplotypes in active regions. Regions showing signs of somatic variation are then reassembled to generate candidate variant haplotypes. Further steps include contamination calculation, orientation bias detection and finally filtering and functional annotation of variants with databases such as GENCODE and dbSNP. You will receive raw (fastq) and aligned (bam) files for each sample, as well as VCF files listing all variants. A bioinformatics report will provide further detail about the tools used and alignment metrics and a variant quality stats.
How to order
Contact us today and one of our skilled account managers will be in touch with a free consultation including further information and pricing details.
Payment options:
Payment can be made by credit card or purchase order number.
FAQ's:
View our frequently asked questions for more information.
Sample requirements:
We require 1-2 ug of gDNA (0D260/280 ratio ranging from 1.8-2) at a minimum concentration of 50 ng/μl and in a minimum volume of 20 μl. Higher concentrations can be submitted and will be diluted accordingly. For full details click here.
How to Order
Contact us today and one of our skilled account managers will be in touch with a free consultation including further information and pricing details.
Next Generation Sequencing
Source BioScience is one of Europe’s leading providers of commercial sequencing, offering Next Generation Sequencing services from our ISO accredited laboratories. We offer NGS services on the most prominent platforms including Illumina’s NovaSeq, NextSeq and MiSeq.